My new blogpost in Kolabtree blog: https://blog.kolabtree.com/applications-of-machine-learning-in-biology/
My new blogpost in Kolabtree blog: https://blog.kolabtree.com/applications-of-machine-learning-in-biology/
This paper was part of my journal club recently. I touched upon LPMOs, short for Lytic Polysaccharide Monooxygenases, in my previous post that are basically oxidative enzymes.
These interesting group of enzymes have three basic types: Type I, Type II, and Type III, classified based on the site of attack, namely LPMO1 (Type I) when oxidation occurs at C1 carbon, LPMO2 (Type II) when oxidation occurs at C4 carbon, and LPMO3 (Type III) if either C1 or C4 carbons are attacked. These subtypes are part of four CAZy families to which LPMOs are categorized into (AA9, AA10, AA11, and AA13).
Having said this, the identification of the types in LPMO is not a trivial task. This specificity to cleave a particular bond, or regiospecificity, is characterized by time-consuming chromatography experiments (HPAEC-PAD), as they are time course studies that involve incubating with the substrate for longer periods. If aldonic acids are discovered in the experiments, then it is C1 cleaving or Type I; and if 4-gemdiol-aldose is detected than it is C4 cleaving of Type II LPMOs.
Given this complex identification protocol, any shortcut to identify the regiospecificity is welcome and that’s what Danneels et al have attempted in their paper published in PLOS ONE. Specifically, using an indicator diagram based identification, they give a solution to identify regiospecificity.
To test they used Hypocrea jecorina‘s LPMO9A (having both C1/C4 cleavage) and did site-directed mutagenesis on key aromatic residues that are involved in substrate binding to create mutants that are either selective to C1 or C4 cleavage. Comparing the activity of the wildtype with the mutants by plotting the speed of release of aldonic acids with respect to 4-gemdiol-aldose the authors plot it as an indicator diagram. Basically, if one calculate the slope of the line, and it is closer to x-axis (release of aldonic acid) then the enzyme’s regiospecificity is for C1 oxidation or consisting of Type I LPMO activity. If closer to y-axis, then Type II LPMO activity.
It would be interesting to see this type of indicator diagram applied for enzyme activity identification for new LPMO enzymes, and also for enzyme engineering studies on LPMO.
Reference: B. Danneels, M. Tanghe, H. Joosten, T. Gundinger, O. Spadiut, I. Stals and T. Desmet, “A quantitative indicator diagram for lytic polysaccharide monooxygenases reveals the role of aromatic surface residues in HjLPMO9A regioselectivity“, 2017. .
Enzyme discovery is always a hot topic for industry and biochemists, since there is huge commercial benefit and advancement in current knowledge-base. Unlike, early days where enzyme discovery relied upon assays, now it is a bioinformatics approach to speed up the process.
Lytic polysaccharide monooxygenases (LPMO) are the latest family of enzymes that have affected the biofuel industry, specifically cellulosic bioethanol industry. These enzymes, previously thought to bind to polymers of carbohydrates, are now understood to boost the enzymatic process of degrading recalcitrant crystalline polysaccharides. LPMOs are probably one of the enzymes that bind to crystalline surfaces of polymers and thus hopefully reducing the cost and time in the pre-processing step of biomass to bioethanol conversion.
As of now, there are 4 families that CAZy identifies to consist of LPMOs (AA9, AA10, AA11, and AA13), where they are now called as Auxillary Activity enzymes. However, there is the possibility that newer families of LPMOs may exist and it depends on how or when we identify them. Voshol et al in their recent paper discuss a bionnformatics based approach and validation using expression data for discovery of novel LPMO families, and report the existence of “LPMO14” family that are active on sugars such as beat-1,3-glucans.
In short, they took 14 known LPMOs from the four families and generated a HMM profile, using which they scanned six genomes and identified 7 LPMO14 genes. They were also able to find LPMO genes in other organisms (such as Drosophila, Bivalves, corals, etc) where the function of LPMO is not known.
While, this data sounds promising, further identification and characterization using standard assays would ensure that this is indeed a new family of LPMOs. Also, the authors do not mention as to why they stuck to 14 known LPMO structures to generate the HMM profile, while there are nearly 50 structures currently deposited in PDB.
Reference: G. Voshol, E. Vijgenboom and P. Punt, “The discovery of novel LPMO families with a new Hidden Markov model“, BMC Research Notes, vol. 10, no. 1, 2017.
The area of protein-protein interactions (PPI) is always exciting as proteins can be either monogamous or promiscuous with their interaction partners. Also, a hot topic these days in computational biology is multiscale modelling. It refers to the method of analyzing a system from atomistic and at a global scale and other scales, in between. Few years ago, a new method of co-evolutionary analysis of residues made news and has been used for gaining insights in other systems. Read the blog by Bosco Ho about the groundbreaking work here about Direct Coupling Analysis (DCA).
Example of a protein-protein interaction. By Dcrjsr – Own work, CC BY 3.0, https://commons.wikimedia.org/w/index.php?curid=10510713
So, when two orthogonal approaches are used to understand a PPI system, it obviously becomes an interesting work. A recent paper in eLife reports exactly that. Malinverni et al report the use of coarse-grained simulation coupled with atomistic molecular dynamics simulation and data from DCA to identify the evolutionarily conserved residues that cause the specific interaction between Hsp70 and Hsp40.
![]() As these two proteins are part of the chaperone machinery, any insight on the Hsp70’s ability to bind to proteins along with Hsp40 is crucial to understand the short-lived interaction. As mentioned in the article, vast amount of data (in terms of NMR, mutagenesis, etc.) is present that can be incorporated in this multiscale modelling.
As these two proteins are part of the chaperone machinery, any insight on the Hsp70’s ability to bind to proteins along with Hsp40 is crucial to understand the short-lived interaction. As mentioned in the article, vast amount of data (in terms of NMR, mutagenesis, etc.) is present that can be incorporated in this multiscale modelling.
The impact of the predicted interaction model is not only statistically significant, but also correlates well with the known experimental data.
Malinverni D, Jost Lopez A, De Los Rios P, Hummer G, Barducci A: Modeling Hsp70/Hsp40 interaction by multi-scale molecular simulations and coevolutionary sequence analysis. eLife 2017, 6. DOI: https://doi.org/10.7554/eLife.23471
Cartoon representation of the molecular structure of protein registered with 2fft code. By Jawahar Swaminathan and MSD staff at the European Bioinformatics Institute – Public Domain, https://commons.wikimedia.org/w/index.php?curid=5877319
Intrinsically disordered proteins are thought to be fully functional, yet do not confirm to a single conformation, thereby identifying their structure via crystallography becomes problematic. Many intrinsically disordered proteins have been studied and analyzed using NMR methods, however the question as to why proteins are intrinsically disordered is still debatable.
While, viewing X-ray diffraction data some residues do not have an electron density region, thus they are marked as missing residues. These regions are highly mobile and are considered as intrinsically disordered. For some proteins, the entire sequence is considered intrinsically disordered.
![]() It is a widely accepted fact that sequence dictates structure, and structure in turn dictates function. So, is the “disordered-ness” encoded in the genome, if so to what extent? This and related questions have led Basile et al at the Stockholm University, Sweden to delve deeper and have narrowed it down to GC content. Their work has been published in latest issue of PLoS Computational Biology.
It is a widely accepted fact that sequence dictates structure, and structure in turn dictates function. So, is the “disordered-ness” encoded in the genome, if so to what extent? This and related questions have led Basile et al at the Stockholm University, Sweden to delve deeper and have narrowed it down to GC content. Their work has been published in latest issue of PLoS Computational Biology.
Using computational methods they analyzed 400 eukaryotic genomes and looked into the so-called orphan genes, specifically. They categorized the age of the proteins using ProteinHistorian tool and looked into the old and young proteins. They found that the
…selective pressure to change amino acids in a protein is stronger than the one to change the GC content. At low GC ancient proteins are more disordered than expected for random sequence while at high GC they are less.
The three disorder promoting amino acids (Ala, Pro, and Gly) are high in GC content w.r.t to their codons. However,
At high GC the youngest proteins become more disordered and contain less secondary structure elements, while at low GC the reverse is observed. We show that these properties can be explained by changes in amino acid frequencies caused by the different amount of GC in different codons.
References:
With increasing computational power (aka GPU) that can be accessed these days, it is no wonder that performing all-atom molecular dynamics simulation for a longer time, with duplicates and/or triplicates, has become easier.
![]()
Two publications report all-atom MD data that have significant implication in two diverse areas. The first one is the popular CRISPR-Cas9 system and the second one is Dengue virus.
With these data it should pave way for more insights.
CRISPR-Cas9 all atom simulation (total of 400-600ns data)
Zuo Z, & Liu J (2016). Cas9-catalyzed DNA Cleavage Generates Staggered Ends: Evidence from Molecular Dynamics Simulations. Scientific reports, 5 PMID: 27874072
Entire Dengue viral envelope complex simluation (1 microsecond data)
Marzinek JK, Holdbrook DA, Huber RG, Verma C, & Bond PJ (2016). Pushing the Envelope: Dengue Viral Membrane Coaxed into Shape by Molecular Simulations. Structure (London, England : 1993), 24 (8), 1410-20 PMID: 27396828
References:
Wanted to share this exciting news with you! Biophysical Journal has created a collection of papers that describe tools and software that can be routinely used in biological research. Editor Prof. Leslie Loew mentions that the full-text of articles in this collection will be freely available until February 25, 2016.
…a year agoBiophysical Journal called for papers in a new class of articles called Computational Tools (CTs). These papers are limited to five pages in length and describe software for analysis of experimental data, modeling and/or simulation, or database services. All are required to be freely accessible and open to the research community. In addition to following the usual review criteria of novelty and importance, reviewers of CTs are asked to test drive the software and judge its usability.
Among the thirteen, some of them are directly related to Structural Biology and Bioinformatics. So, here’s my “curated” list.
![]()
Article Title
CHARMM-GUI HMMM Builder for Membrane Simulations with the Highly Mobile Membrane-Mimetic Model
Weblink: http://www.charmm-gui.org/input/hmmm
Abstract
Slow diffusion of the lipids in conventional all-atom simulations of membrane systems makes it difficult to sample large rearrangements of lipids and protein-lipid interactions. Recently, Tajkhorshid and co-workers developed the highly mobile membrane-mimetic (HMMM) model with accelerated lipid motion by replacing the lipid tails with small organic molecules. The HMMM model provides accelerated lipid diffusion by one to two orders of magnitude, and is particularly useful in studying membrane-protein associations. However, building an HMMM simulation system is not easy, as it requires sophisticated treatment of the lipid tails. In this study, we have developed CHARMM-GUI HMMM Builder (http://www.charmm-gui.org/input/hmmm) to provide users with ready-to-go input files for simulating HMMM membrane systems with/without proteins. Various lipid-only and protein-lipid systems are simulated to validate the qualities of the systems generated by HMMM Builder with focus on the basic properties and advantages of the HMMM model. HMMM Builder supports all lipid types available in CHARMM-GUI and also provides a module to convert back and forth between an HMMM membrane and a full-length membrane. We expect HMMM Builder to be a useful tool in studying membrane systems with enhanced lipid diffusion.
Article Title
MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories
Weblink: http://mdtraj.org/latest/
Abstract
As molecular dynamics (MD) simulations continue to evolve into powerful computational tools for studying complex biomolecular systems, the necessity of flexible and easy-to-use software tools for the analysis of these simulations is growing. We have developed MDTraj, a modern, lightweight, and fast software package for analyzing MD simulations. MDTraj reads and writes trajectory data in a wide variety of commonly used formats. It provides a large number of trajectory analysis capabilities including minimal root-mean-square-deviation calculations, secondary structure assignment, and the extraction of common order parameters. The package has a strong focus on interoperability with the wider scientific Python ecosystem, bridging the gap between MD data and the rapidly growing collection of industry-standard statistical analysis and visualization tools in Python. MDTraj is a powerful and user-friendly software package that simplifies the analysis of MD data and connects these datasets with the modern interactive data science software ecosystem in Python.
Article Title
MDN: A Web Portal for Network Analysis of Molecular Dynamics Simulations
Weblink: http://mdn.cheme.columbia.edu/
Abstract
We introduce a web portal that employs network theory for the analysis of trajectories from molecular dynamics simulations. Users can create protein energy networks following methodology previously introduced by our group, and can identify residues that are important for signal propagation, as well as measure the efficiency of signal propagation by calculating the network coupling. This tool, called MDN, was used to characterize signal propagation in Escherichia coli heat-shock protein 70-kDa. Two variants of this protein experimentally shown to be allosterically active exhibit higher network coupling relative to that of two inactive variants. In addition, calculations of partial coupling suggest that this quantity could be used as part of the criteria to determine pocket druggability in drug discovery studies.
Article Title
Multidomain Assembler (MDA) Generates Models of Large Multidomain Proteins
Weblink: http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/mda.html AND http://www.cell.com/biophysj/biophysj/supplemental/S0006-3495(15)00339-2
Abstract
Homology modeling predicts protein structures using known structures of related proteins as templates. We developed MULTIDOMAIN ASSEMBLER (MDA) to address the special problems that arise when modeling proteins with large numbers of domains, such as fibronectin with 30 domains, as well as cases with hundreds of templates. These problems include how to spatially arrange nonoverlapping template structures, and how to get the best template coverage when some sequence regions have hundreds of available structures while other regions have a few distant homologs. MDA automates the tasks of template searching, visualization, and selection followed by multidomain model generation, and is part of the widely used molecular graphics package UCSF CHIMERA (University of California, San Francisco). We demonstrate applications and discuss MDA’s benefits and limitations.
Article Title
RedMDStream: Parameterization and Simulation Toolbox for Coarse-Grained Molecular Dynamics Models
Weblink: https://bionano.cent.uw.edu.pl/Software/RedMD
Abstract
Coarse-grained (CG) models in molecular dynamics (MD) are powerful tools to simulate the dynamics of large biomolecular systems on micro- to millisecond timescales. However, the CG model, potential energy terms, and parameters are typically not transferable between different molecules and problems. So parameterizing CG force fields, which is both tedious and time-consuming, is often necessary. We present RedMDStream, a software for developing, testing, and simulating biomolecules with CG MD models. Development includes an automatic procedure for the optimization of potential energy parameters based on metaheuristic methods. As an example we describe the parameterization of a simple CG MD model of an RNA hairpin.
Article Title
A Web Interface for Easy Flexible Protein-Protein Docking with ATTRACT
Weblink: http://www.attract.ph.tum.de/services/ATTRACT/attract.html
Abstract
Protein-protein docking programs can give valuable insights into the structure of protein complexes in the absence of an experimental complex structure. Web interfaces can facilitate the use of docking programs by structural biologists. Here, we present an easy web interface for protein-protein docking with the ATTRACT program. While aimed at nonexpert users, the web interface still covers a considerable range of docking applications. The web interface supports systematic rigid-body protein docking with the ATTRACT coarse-grained force field, as well as various kinds of protein flexibility. The execution of a docking protocol takes up to a few hours on a standard desktop computer.
Article Title
ReaDDyMM: Fast Interacting Particle Reaction-Diffusion Simulations Using Graphical Processing Units
Weblink: https://github.com/readdy
Abstract
ReaDDy is a modular particle simulation package combining off-lattice reaction kinetics with arbitrary particle interaction forces. Here we present a graphical processing unit implementation of ReaDDy that employs the fast multiplatform molecular dynamics package OpenMM. A speedup of up to two orders of magnitude is demonstrated, giving us access to timescales of multiple seconds on single graphical processing units. This opens up the possibility of simulating cellular signal transduction events while resolving all protein copies.
Article Title
Local Perturbation Analysis: A Computational Tool for Biophysical Reaction-Diffusion Models
Weblink: http://www.cell.com/biophysj/biophysj/supplemental/S0006-3495(14)04670-0
Abstract
Diffusion and interaction of molecular regulators in cells is often modeled using reaction-diffusion partial differential equations. Analysis of such models and exploration of their parameter space is challenging, particularly for systems of high dimensionality. Here, we present a relatively simple and straightforward analysis, the local perturbation analysis, that reveals how parameter variations affect model behavior. This computational tool, which greatly aids exploration of the behavior of a model, exploits a structural feature common to many cellular regulatory systems: regulators are typically either bound to a membrane or freely diffusing in the interior of the cell. Using well-documented, readily available bifurcation software, the local perturbation analysis tracks the approximate early evolution of an arbitrarily large perturbation of a homogeneous steady state. In doing so, it provides a bifurcation diagram that concisely describes various regimes of the model’s behavior, reducing the need for exhaustive simulations to explore parameter space. We explain the method and provide detailed step-by-step guides to its use and application.
References:
We all have neighbors who help us in our hour of need. Some go out of the way as well. In enzymes too, it seems, that neighbors play a crucial role. Lafond et al in their recent publication in the Journal of Biological Chemistry report the invovlement of neighboring chains of the same enzyme, lichenase. Apart from the role of stabilizing the quarternary structure (a trimer), they are also invovled in the enzymatic activity.
Sacchrophagus degradans is a marine bacteria that has been credited with the capacity of degrading diverse polysaccharides substrates. The list includes, but not limited to, agar, cellulose, chitin, xylan, carboxymethylcellulose, avicel, laminarin, wheat arabinoxylan, glucomannan, lichenan, curdlan, pachyman, and others. Its genome has 19 coding regions for enzymes that belong to the same CAZy family called GH5.
![]() GH5 class of enzymes are predominantly endoglucanases, i.e. cleave an internal beta-glycosidic bond in the cellulose polymer. They are also characterized by sharing the same protein structural fold, namely the (alpha/beta)8 fold. There are eight beta strands with alternating helices forming a barrel. The enzyme Lafond et al named SdGluc5_26A, also belongs to GH5 family with the classical (alpha/beta)8 fold. However, they also found a stretch of 38 residues at the N terminus that seemed interesting. This N-terminus is not floppy, but binds to the active site of the neighboring chain.
GH5 class of enzymes are predominantly endoglucanases, i.e. cleave an internal beta-glycosidic bond in the cellulose polymer. They are also characterized by sharing the same protein structural fold, namely the (alpha/beta)8 fold. There are eight beta strands with alternating helices forming a barrel. The enzyme Lafond et al named SdGluc5_26A, also belongs to GH5 family with the classical (alpha/beta)8 fold. However, they also found a stretch of 38 residues at the N terminus that seemed interesting. This N-terminus is not floppy, but binds to the active site of the neighboring chain.
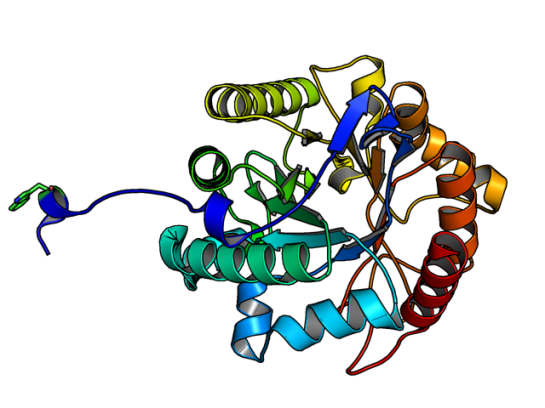
Image of SdGluc5_26A made using PyMOL. (PDB id: 5a8n)
In the figure above, the Trp residue (shown in green sticks) specifically binds to the active site of the neighboring chain. See the figure of the trimer below to see how they interact. Such an arrangement made SdGluc5_26A behave with lichenase activity. In the parlance of carbohydrate active enzymes, this Trp was binding to the -3 subsite of the active site.
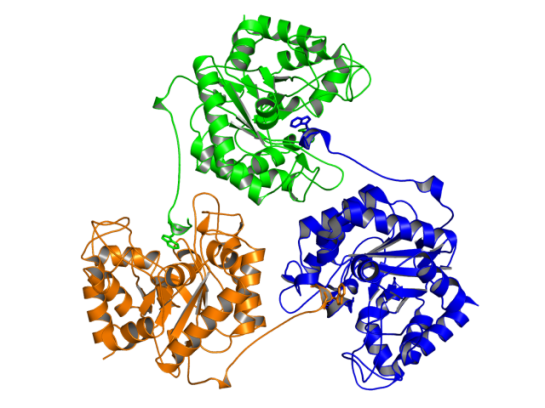
Image of SdGluc5_26A trimer made using PyMOL. (PDB id: 5a8n)
So, the next step was to find out what happened to the activity of SdGluc5_26A, when this protruding N-terminal sequence is deleted. It was observed that upon deletion, SdGluc5_26A now behaved as a endo-beta(1,4)-glucanase. In other words, without this N-terminal part the enzyme switched its activity from an exo (chewing at the ends of the polymer) to an endo (chewing in the middle) reactive enzyme.
Given that SdGluc5_26A can act on variety of substrates, it only logical to think that this 38 residue stretch plays an important role in substrate specificity. Now, the question is if there is any allostery and cooperative mechanism that can be the reason for substrate binding? Something to chew upon! 😉
References:

The Mosquito Net (1912) by John Singer Sargent. Licensed under Public Domain via Wikimedia Commons
We all know how pesky mosquitoes can be. Did you know that the ability of a mosquito to find a suitable host to feed is due to thermotaxis? This behavior, being attracted/repelled due to high/low temperature, is seen in other organisms as well such as Drosophila melanogaster and Caenorhabditis elegans.
However, the behaviour is more pronounced among blood-feeding pests (kissing bugs, bedbugs, Ticks, and mosquitoes including Aedes aegypti). Aedes aegypti is a vector for many flaviviral diseases (Dengue fever, Yellow fever, etc.) Until now, it was well established that thermotaxis requires specific thermosensors that activate the sensory signals for a subsequent flight response in a mosquito. However, how exactly they function was not resolved.
![]() In a recent paper by Corfas and Vosshall [1] describe the use of zinc-finger nuclease-mediated genome editing method to identify the role of two receptors TRPA1 and GR19 in Aedes aegypti‘s attraction to heat. It was found that these receptors help the mosquito to identify the host for feeding (in the temperature range of 43-50 deg Celcius), however they avoid surfaces that exhibit above 50 deg Celcius. [Read the recent editorial on genome editing in Genome Biology]
In a recent paper by Corfas and Vosshall [1] describe the use of zinc-finger nuclease-mediated genome editing method to identify the role of two receptors TRPA1 and GR19 in Aedes aegypti‘s attraction to heat. It was found that these receptors help the mosquito to identify the host for feeding (in the temperature range of 43-50 deg Celcius), however they avoid surfaces that exhibit above 50 deg Celcius. [Read the recent editorial on genome editing in Genome Biology]
The sequence (923 residues long) of this receptor (Uniprot id: Q0IFQ4) has at least five transmembrane regions that are approximately 20-25 residues long. A cursory glance at homologous sequences shows that it shares 37% sequence identity with the a de novo designed protein (PDB id:2xeh).
The homology modeled structure showing coiled coil region (residues 189-338). Although, the eLife paper does not talk about structure, I felt that this paper deserves a mention here. The reason is the structural biology/bioinformatics possibilities with this novel target. It is a suitable target for designing inhibitors that would potentially act as mosquito repellents.
Also, combined with the method described in my previous post on mutating transmembrane proteins as a method of making them crystallize, I guess the 3D structure of this important protein will come to light sooner!
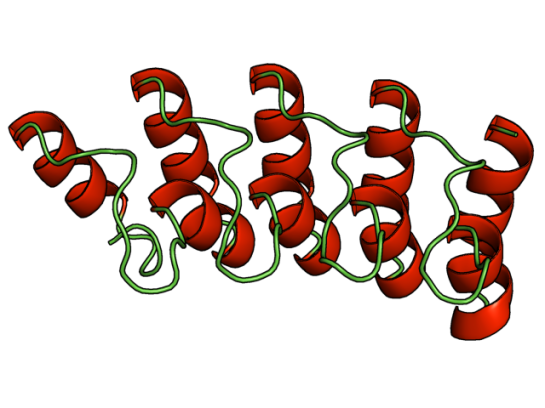
Homology modeled region of TRPA1 (189-338), from ModBase
References:
I am sharing this guest post of mine that was published in Cell’s Crosstalk: Biology in 3D Blog. Yes, the journal Cell!
Here is the link: http://www.cell.com/crosstalk/why-do-i-blog-about-structural-bioinformatics
Enjoy!

When someone says that they have a blog, the stereotypical response would be, “About your travels?” or “Hmmm … Recipes … Must be a delicious blog!” And when one confesses that said blog is about scientific research, the jaw drops. I presume it has to do with the notion that blogging science is not that much fun!
Two things inspired me to become a blogger: (1) an amazing community of scientific bloggers at Research Blogging, who inspired me with their wonderful posts; and (2) my view that structural biology and structural bioinformatics are not getting the exposure they deserve. Thus inspired and motivated, I begun blogging about four years ago, and was able to channel some of my thoughts and energy into my blog, called Getting to Know Structural Bioinformatics.
 |
|
Guest author and blogger |
Why do I blog? Blogging is fun! For me, blogging is about sharing with the world recent research and tidbits on structural biology and bioinformatics. Most importantly, it is about sharing the excitement that I feel after reading a paper. In some sense, blogging about research is similar to a journal club, where I am able to share the latest research with my peers. However, unlike a journal club, the audience for my blog is the entire world.
Blogging is also dynamic and interactive, because it allows me to engage in conversation with others (specifically students) when they weigh in with their comments. Below I highlight some of the best practices that I’ve developed over the years that help me with balancing my research, teaching, and personal responsibilities with my blogging.
The main way I find articles that I want to blog about is by scouring through the table of contents of the journals I am interested in. Sometimes I also hear about exciting protein structures via friends and other blogs that I follow. I try to have a balanced approach and highlight structural work on systems that are “hot topics” as well as papers that just captured my interest and fancy.
In the early days of my blogging, I was trying to collate and compile tools and techniques that would come in handy for students working with protein structures. I wanted my blog to be a handy place for myself and others to find tips and tricks. Over time, the range of topics and papers I cover has broadened, and although I still cover a lot of method development work, I cover other topics as well. In general, once I make up my mind about the paper I want to blog about, I start reading it, give myself some time to soak in the method and outcome of the paper, and try to think critically as to what possible gaps or methods that the authors could have done to make the paper better. Alternatively, I also analyze the paper’s novelty with respect to structural bioinformatics.
I should confess that the monthly posts in Protein Spotlight by Vivienne Baillie Gerritsen are my inspiration while composing posts. I love her writing style and also the manner in which artwork is included in every post, to make it fun to read. Like Protein Spotlight, blogs have the advantage of including other multimedia items, for example using animated gifs and YouTube videos that make the post much easier for the reader. So, I start finding an appropriate image from an art database that best fits the topic (of course, giving credit where it is due). When it is about a tool/software, I figure the best approach is to use said tool/software and include a “first-hand” experience of how I perceived it. Also, I try to include an additional tidbit or information that the authors mention in passing.
With an active teaching and research schedule, finding time to blog does become a challenge. I try to make it a fun process, so that it does not feel cumbersome. If one looks at the frequency of my posts, I try to maintain at least one post per month. Looking at others’ blogs at Research Blogging, I realize that one post a month is a low turnout, and I try to post as frequently as possible. Sometimes, the problem is sheer lack of time or not finding exciting enough material to blog about. However, this does not mean that exciting research is not out there. The key is to find a balance between blogging and other duties. I have had discussions with other bloggers who blog on other nonscience topics, and we observed that the main turnoff in blogging is when one delves deeper and over time a particular post becomes “work.” Maneuvering that roadblock is key to maintaining a successful blog.
In the end, as at the beginning, it all comes down to having fun and sharing with the world my excitement about the type of scientific research I enjoy. I think this is probably the feeling others who blog share as well, and I can see it in some of the blogs I follow, such as the following:
Raghu Yennamalli completed his PhD in Computational Biology and Bioinformatics in 2008 from Jawaharlal Nehru University. He conducted postdoctoral research at Iowa State University, University of Wisconsin-Madison, and Rice University. Currently, he is an Assistant Professor at Jaypee University of Information Technology. He can be contacted at ragothaman AT gmail DOT com.
