We all have neighbors who help us in our hour of need. Some go out of the way as well. In enzymes too, it seems, that neighbors play a crucial role. Lafond et al in their recent publication in the Journal of Biological Chemistry report the invovlement of neighboring chains of the same enzyme, lichenase. Apart from the role of stabilizing the quarternary structure (a trimer), they are also invovled in the enzymatic activity.
Sacchrophagus degradans is a marine bacteria that has been credited with the capacity of degrading diverse polysaccharides substrates. The list includes, but not limited to, agar, cellulose, chitin, xylan, carboxymethylcellulose, avicel, laminarin, wheat arabinoxylan, glucomannan, lichenan, curdlan, pachyman, and others. Its genome has 19 coding regions for enzymes that belong to the same CAZy family called GH5.
![]() GH5 class of enzymes are predominantly endoglucanases, i.e. cleave an internal beta-glycosidic bond in the cellulose polymer. They are also characterized by sharing the same protein structural fold, namely the (alpha/beta)8 fold. There are eight beta strands with alternating helices forming a barrel. The enzyme Lafond et al named SdGluc5_26A, also belongs to GH5 family with the classical (alpha/beta)8 fold. However, they also found a stretch of 38 residues at the N terminus that seemed interesting. This N-terminus is not floppy, but binds to the active site of the neighboring chain.
GH5 class of enzymes are predominantly endoglucanases, i.e. cleave an internal beta-glycosidic bond in the cellulose polymer. They are also characterized by sharing the same protein structural fold, namely the (alpha/beta)8 fold. There are eight beta strands with alternating helices forming a barrel. The enzyme Lafond et al named SdGluc5_26A, also belongs to GH5 family with the classical (alpha/beta)8 fold. However, they also found a stretch of 38 residues at the N terminus that seemed interesting. This N-terminus is not floppy, but binds to the active site of the neighboring chain.
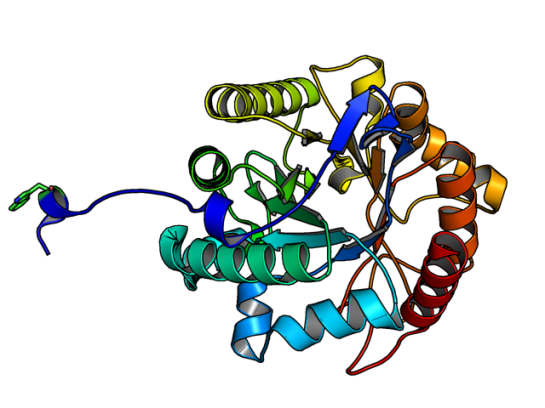
Image of SdGluc5_26A made using PyMOL. (PDB id: 5a8n)
In the figure above, the Trp residue (shown in green sticks) specifically binds to the active site of the neighboring chain. See the figure of the trimer below to see how they interact. Such an arrangement made SdGluc5_26A behave with lichenase activity. In the parlance of carbohydrate active enzymes, this Trp was binding to the -3 subsite of the active site.
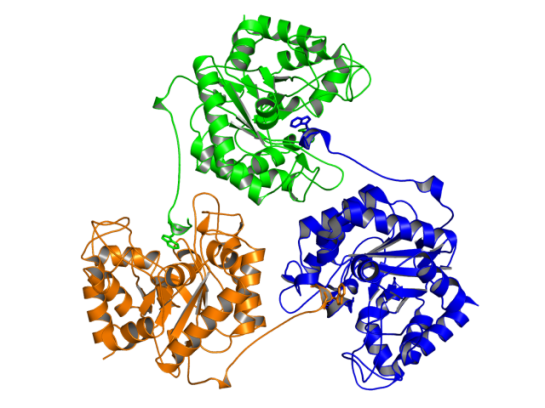
Image of SdGluc5_26A trimer made using PyMOL. (PDB id: 5a8n)
So, the next step was to find out what happened to the activity of SdGluc5_26A, when this protruding N-terminal sequence is deleted. It was observed that upon deletion, SdGluc5_26A now behaved as a endo-beta(1,4)-glucanase. In other words, without this N-terminal part the enzyme switched its activity from an exo (chewing at the ends of the polymer) to an endo (chewing in the middle) reactive enzyme.
Given that SdGluc5_26A can act on variety of substrates, it only logical to think that this 38 residue stretch plays an important role in substrate specificity. Now, the question is if there is any allostery and cooperative mechanism that can be the reason for substrate binding? Something to chew upon! 😉
References:
- Lafond M, Sulzenbacher G, Freyd T, Henrissat B, Berrin JG, & Garron ML (2016). the quaternary structure of a glycoside hydrolase dictates specificity towards beta-glucans. The Journal of biological chemistry PMID: 26755730